Annotations¶
Annotations help users search for and find data, and they are a powerful tool used to systematically group and/or describe things in Synapse.
Annotations are stored as key-value pairs in Synapse, where the key defines a particular aspect of your data, for example (species, assay, fileFormat) and the value defines a variable that belongs to that category (mouse, RNAseq, .bam). You can use annotations to add additional information about a project, file, folder, table, or view.
Annotations can be based on an existing ontology or controlled vocabulary, or can be created as needed and modified later as your metadata evolves.
Note: You may optionally follow the Uploading data in bulk tutorial instead. The bulk tutorial may fit your needs better as it limits the amount of code that you are required to write and maintain.
Tutorial Purpose¶
In this tutorial you will:
- Add several annotations to stored files
- Upload 2 new files and set the annotations at the same time
Prerequisites¶
- Make sure that you have completed the File tutorial or have at least 1 file in your Synapse project.
1. Add several annotations to stored files¶
First let's retrieve all of the Synapse IDs we are going to use¶
# Step 1: Add several annotations to stored files
import os
import synapseclient
from synapseclient import File
syn = synapseclient.login()
# Retrieve the project ID
my_project_id = syn.findEntityId(
name="My uniquely named project about Alzheimer's Disease"
)
# Retrieve the folders I want to annotate files in
batch_1_folder_id = syn.findEntityId(
name="single_cell_RNAseq_batch_1", parent=my_project_id
)
Next let's define the annotations I want to set¶
# Define the annotations I want to set
annotation_values = {
"species": "Homo sapiens",
"dataType": "geneExpression",
"assay": "SCRNA-seq",
"fileFormat": "fastq",
Finally we'll loop over all of the files and set their annotations¶
# Loop over all of the files and set their annotations
for file_batch_1 in syn.getChildren(parent=batch_1_folder_id, includeTypes=["file"]):
# Grab and print the existing annotations this File may already have
existing_annotations_for_file = syn.get_annotations(entity=file_batch_1)
print(
f"Got the annotations for File: {file_batch_1['name']}, ID: {file_batch_1['id']}, Annotations: {existing_annotations_for_file}"
)
# Merge the new annotations with anything existing
existing_annotations_for_file.update(annotation_values)
existing_annotations_for_file = syn.set_annotations(
annotations=existing_annotations_for_file
)
print(
f"Set the annotations for File: {file_batch_1['name']}, ID: {file_batch_1['id']}, Annotations: {existing_annotations_for_file}"
You'll see that each file now has a number of annotations:
Batch 1 Folder ID: syn53205629
Got the annotations for File: SRR12345678_R1.fastq.gz, ID: syn53205687, Annotations: {}
Set the annotations for File: SRR12345678_R1.fastq.gz, ID: syn53205687, Annotations: {'assay': ['SCRNA-seq'], 'species': ['Homo sapiens'], 'dataType': ['geneExpression'], 'fileFormat': ['fastq']}
Got the annotations for File: SRR12345678_R2.fastq.gz, ID: syn53205688, Annotations: {}
Set the annotations for File: SRR12345678_R2.fastq.gz, ID: syn53205688, Annotations: {'assay': ['SCRNA-seq'], 'species': ['Homo sapiens'], 'dataType': ['geneExpression'], 'fileFormat': ['fastq']}
2. Upload 2 new files and set the annotations at the same time¶
Assuming we have a few new files we want to upload we'll follow a similar pattern defined
in the File tutorial, except now we'll specify the annotations attribute before
uploading the file to Synapse.
In order for the following script to work please replace the files with ones that already exist on your local machine.
# Step 2: Upload 2 new files and set the annotations at the same time
# In order for the following script to work please replace the files with ones that
# already exist on your local machine.
batch_1_scrnaseq_new_file_1 = File(
path=os.path.expanduser(
"~/my_ad_project/single_cell_RNAseq_batch_1/SRR92345678_R1.fastq.gz"
),
parent=batch_1_folder_id,
annotations=annotation_values,
)
batch_1_scrnaseq_new_file_2 = File(
path=os.path.expanduser(
"~/my_ad_project/single_cell_RNAseq_batch_1/SRR92345678_R2.fastq.gz"
),
parent=batch_1_folder_id,
annotations=annotation_values,
)
batch_1_scrnaseq_new_file_1 = syn.store(obj=batch_1_scrnaseq_new_file_1)
batch_1_scrnaseq_new_file_2 = syn.store(obj=batch_1_scrnaseq_new_file_2)
print(
f"Stored file: {batch_1_scrnaseq_new_file_1['name']}, ID: {batch_1_scrnaseq_new_file_1['id']}, Annotations: {batch_1_scrnaseq_new_file_1['annotations']}"
)
print(
f"Stored file: {batch_1_scrnaseq_new_file_2['name']}, ID: {batch_1_scrnaseq_new_file_2['id']}, Annotations: {batch_1_scrnaseq_new_file_2['annotations']}"
You'll notice the output looks like:
Stored file: SRR92345678_R1.fastq.gz, ID: syn53206218, Annotations: {
"assay": [
"SCRNA-seq"
],
"dataType": [
"geneExpression"
],
"fileFormat": [
"fastq"
],
"species": [
"Homo sapiens"
]
}
Stored file: SRR92345678_R2.fastq.gz, ID: syn53206219, Annotations: {
"assay": [
"SCRNA-seq"
],
"dataType": [
"geneExpression"
],
"fileFormat": [
"fastq"
],
"species": [
"Homo sapiens"
]
}
Results¶
Now that you have annotated your files you'll be able to inspect this on the individual files in the synapse web UI. It should look similar to:
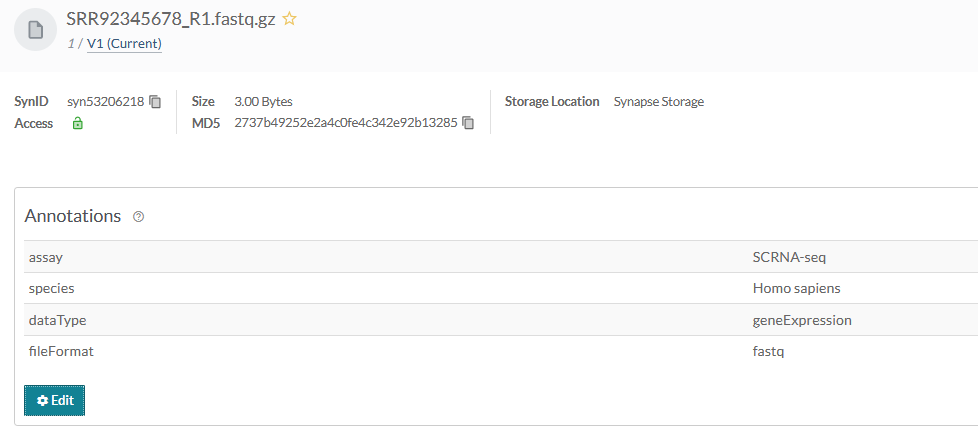
Source code for this tutorial¶
Click to show me
"""
Here is where you'll find the code for the Annotation tutorial.
"""
# Step 1: Add several annotations to stored files
import os
import synapseclient
from synapseclient import File
syn = synapseclient.login()
# Retrieve the project ID
my_project_id = syn.findEntityId(
name="My uniquely named project about Alzheimer's Disease"
)
# Retrieve the folders I want to annotate files in
batch_1_folder_id = syn.findEntityId(
name="single_cell_RNAseq_batch_1", parent=my_project_id
)
print(f"Batch 1 Folder ID: {batch_1_folder_id}")
# Define the annotations I want to set
annotation_values = {
"species": "Homo sapiens",
"dataType": "geneExpression",
"assay": "SCRNA-seq",
"fileFormat": "fastq",
}
# Loop over all of the files and set their annotations
for file_batch_1 in syn.getChildren(parent=batch_1_folder_id, includeTypes=["file"]):
# Grab and print the existing annotations this File may already have
existing_annotations_for_file = syn.get_annotations(entity=file_batch_1)
print(
f"Got the annotations for File: {file_batch_1['name']}, ID: {file_batch_1['id']}, Annotations: {existing_annotations_for_file}"
)
# Merge the new annotations with anything existing
existing_annotations_for_file.update(annotation_values)
existing_annotations_for_file = syn.set_annotations(
annotations=existing_annotations_for_file
)
print(
f"Set the annotations for File: {file_batch_1['name']}, ID: {file_batch_1['id']}, Annotations: {existing_annotations_for_file}"
)
# Step 2: Upload 2 new files and set the annotations at the same time
# In order for the following script to work please replace the files with ones that
# already exist on your local machine.
batch_1_scrnaseq_new_file_1 = File(
path=os.path.expanduser(
"~/my_ad_project/single_cell_RNAseq_batch_1/SRR92345678_R1.fastq.gz"
),
parent=batch_1_folder_id,
annotations=annotation_values,
)
batch_1_scrnaseq_new_file_2 = File(
path=os.path.expanduser(
"~/my_ad_project/single_cell_RNAseq_batch_1/SRR92345678_R2.fastq.gz"
),
parent=batch_1_folder_id,
annotations=annotation_values,
)
batch_1_scrnaseq_new_file_1 = syn.store(obj=batch_1_scrnaseq_new_file_1)
batch_1_scrnaseq_new_file_2 = syn.store(obj=batch_1_scrnaseq_new_file_2)
print(
f"Stored file: {batch_1_scrnaseq_new_file_1['name']}, ID: {batch_1_scrnaseq_new_file_1['id']}, Annotations: {batch_1_scrnaseq_new_file_1['annotations']}"
)
print(
f"Stored file: {batch_1_scrnaseq_new_file_2['name']}, ID: {batch_1_scrnaseq_new_file_2['id']}, Annotations: {batch_1_scrnaseq_new_file_2['annotations']}"
)